Aniridia: příčiny, příznaky, léčba, co to je, pravděpodobnost zdědění nepřítomnosti duhovky
úvod: Kongenitální aniridie (CA) je monogenní dědičná patologie, která se vyskytuje v populaci s frekvencí 1:57. WA se vyskytuje jako izolovaná patologie: sporadická nebo familiární s autozomálně dominantním typem dědičnosti a jako součást syndromu WAGR (~143 %). Hlavními diagnostickými příznaky ZO jsou vrozená absence duhovky a hypoplazie fovey, doprovázená nystagmem, navíc u většiny pacientů jsou postiženy i jiné struktury oka a mohou být pozorovány morfologické a funkční poruchy mozku. V současné době nebyly stanoveny žádné specifické vztahy mezi typem mutace genu PAX13 (chromozomální delece) a klinickými rysy VA.
účel práce spočívá v analýze vzorců spojujících genotyp pacientů pro gen PAX6 s klinickými projevy VA.
Materiál a metody: Analýza zahrnovala 98 pacientů s VA ze 73 nepříbuzných rodin s malými mutacemi v genu PAX6 (74 pacientů) a velkými chromozomálními delecemi 11p13 (24 pacientů). Byly vytvořeny skupiny pacientů s genotypy podobnými v molekulárních důsledcích nebo v souladu s lokalizací mutace. Mezi typem/místem mutace a znakem VA byly sestaveny kontingenční tabulky 2×2. Statistická analýza byla provedena pomocí Fisherova exaktního testu.
výsledky: Bylo zjištěno, že fenotypy VA spojené s mutacemi se ztrátou funkce (nesmyslné mutace, posunové mutace a sestřihové mutace) jsou charakterizovány závažnějším klinickým průběhem. Missense mutace jsou spojeny s fenotypem, který se neliší od toho v obecném vzorku, ale parciální aniridie se vyskytuje významně častěji u missense mutací. Fenotypy pacientů s chromozomálními delecemi, s výjimkou delecí 3′ cis-regulační oblasti, se neliší od fenotypů pacientů s intragenními mutacemi PAX6. Mírnější fenotyp je pozorován u pacientů s delecemi 3′ cis-regulační oblasti genu PAX6.
Klíčová slova: vrozená aniridie, mutace genu PAX6, chromozomální delece oblasti 11p13, vztah mezi typem mutace a klinickými znaky.
Klinické a molekulárně-genetické rysy vrozené aniridie
Vasiljeva TA 1, Voskresenskaya AA 2, Kadyshev VV 1, Pozdějeva NA 2, Marakhonov AV 1,3, Zinchenko RA 1,4
1 Výzkumné centrum lékařské genetiky, Moskva
2 Čeboksary pobočka S. Fjodorova oční mikrochirurgie Federální státní instituce
3 Moskevský fyzikální a technologický institut (Státní univerzita), Dolgoprudny
4 Pirogov Ruská lékařská univerzita národního výzkumu, Moskva
Úvod: kongenitální aniridie (AN) je mendelovská autozomálně dominantní porucha (populační prevalence 1:57143). AN se může objevit jako součást syndromu WAGR (~13 %). Hlavními diagnostickými příznaky AN jsou absence hypoplazie duhovky a fovey doprovázená nystagmem. Pacienti také vykazují jiné oční struktury a také anomálie centrálního nervového systému. AN je způsobena heterozygotními mutacemi genu PAX6 nebo chromozomovými přestavbami oblasti 11p13. Neexistuje žádná prokázaná korelace mezi typem mutace PAX6 a rysy klinického obrazu aniridie.
Cíl: práce má analyzovat pravděpodobné vztahy mezi klinickými příznaky AN a typem mutace PAX6.
Pacienti a metody: Bylo analyzováno 98 pacientů s AN ze 73 nepříbuzných rodin s identifikovanými malými mutacemi PAX6 (74 pacientů) a velkými chromozomálními delecemi 11p13 (24 pacientů). Pacienti byli rozděleni do skupin podle typu/místa mutace. Fenotypové znaky byly odkazovány na typ/umístění mutace. Kontingenční tabulky 2X2 byly analyzovány přesným Fisherovým testem.
výsledky: AN klinický obraz spojený se ztrátou funkčních mutací (nesmysl, posun rámce a sestřih) má závažnější klinický průběh. Missense mutace jsou spojeny s fenotypem charakteru obecného vzorku, nicméně parciální aniridie se vyskytuje podstatně častěji. Fenotypy pacientů s chromozomálními delecemi (bez delecí 3′ cis-regulační oblasti) se neliší od pacientů s intragenními mutacemi. Mírnější fenotyp je pozorován u pacientů s delecemi 3′ cis-regulační oblasti.
Klíčová slova: vrozená aniridie, mutace PAX6, delece oblasti chromozomu 11p13, vztahy mezi typem mutace a klinickým znakem.
Citace: Vasilyeva TA, Voskresenskaya AA, Kadyshev VV vůbec. Klinické a molekulárně-genetické rysy kongenitální aniridie // RMJ „Klinická oftalmologie“. 2018;1:7–12.
Klíčová slova:
Článek je věnován studiu klinických, molekulárních a genetických rysů kongenitálních aniridií. Klinické příznaky kongenitální aniridie byly identifikovány v závislosti na detekovaných mutacích.
úvod
Identifikace vztahu mezi klinickými znaky fenotypu a genotypem potvrzeným molekulárně genetickými metodami umožňuje nejen objasnit diferenciální diagnostiku onemocnění, ale také rozšiřuje naše chápání klinického průběhu onemocnění a koriguje možnou léčbu.
Kongenitální aniridie (OMIM #106210) (CA) je monogenní dědičná patologie s celosvětovou prevalencí v populacích po celém světě, podle registru vzácných a sirotčích onemocnění Orphanet, 1:57 143 populace [1]. Hlavními diagnostickými příznaky jsou vrozená absence tkáně duhovky, hypoplazie fovey, doprovázená nystagmem [2].
Ve většině případů se VA vyskytuje jako izolovaná patologie: sporadická nebo familiární s autozomálně dominantním typem dědičnosti (85 %) a syndromická (15 %). Syndromová WA je detekována ve 13 % případů jako součást WAGR syndromu (OMIM #194072) a ve 2 % případů jsou pozorovány atypické formy WA s recesivní dědičností [2]. WAGR syndrom je charakterizován 4 klinickými příznaky: Wilmsův tumor, aniridie, genitourinární anomálie a mentální retardace [3].
Ve většině případů je VA způsobena heterozygotními mutacemi v genu PAX6 (OMIM*607108) [4], lokalizovaným na 11. chromozomu, včetně chromozomálních přestaveb (oblast 11p13) [5]. WAGR syndrom je způsoben delecemi oblasti 11p13 zahrnující lokusy genu PAX6 a genu vnímavosti Wilmsových nádorů WT1 (OMIM*607102).
Variabilita klinického obrazu může být velmi vysoká. Kromě hlavních příznaků se může vyvinout VA: opacity a/nebo subluxace čočky různého stupně, keratopatie (u 80 % pacientů) a méně častý glaukom a hypoplazie zrakového nervu. U VA je pozorován významný pokles zrakové ostrosti [6]. V 85 % případů izolované ZO jsou tedy kromě duhovky postiženy i další struktury oka. V 10 % případů izolované WA mohou být malformace očních struktur provázeny lézemi centrálního nervového, endokrinního, genitourinárního a dalších systémů a orgánů, které se liší od známek syndromu WAGR [7].
V současné době nebyly stanoveny žádné specifické vztahy mezi typem mutace genu PAX6 ani přítomností a velikostí chromozomální delece oblasti 11p13 a charakteristikou klinického obrazu VA včetně její závažnosti [8].
Cílem této práce je hledání možných genotypových korelací mezi klinickým obrazem onemocnění a genotypem stanoveným při konfirmační diagnostice.
materiály a metody
Výzkumný tým Laboratoře genetické epidemiologie Federálního státního rozpočtového vědeckého ústavu „Výzkumné centrum lékařské genetiky“ jako první v Rusku provedl molekulárně genetickou analýzu 110 pacientů z 84 nepříbuzných rodin s presumptivní diagnózou „vrozená aniridie“ [9].
Konfirmační a diferenciální diagnostika ZO byla prováděna v souladu s protokolem dříve vyvinutým v laboratoři genetické epidemiologie Federálního státního rozpočtového vědeckého ústavu „Výzkumné centrum lékařské genetiky“ [10, 11]. Všechny subjekty daly informovaný souhlas se zpracováním osobních údajů a klinickým laboratorním vyšetřením, studie byla schválena Etickou komisí Federálního státního rozpočtového vědeckého ústavu „Výzkumné centrum lékařské genetiky“.
Kritéria pro zařazení pacientů do této analýzy genotypových korelací byla:
klinická diagnóza VA;
prokázaná molekulární příčina onemocnění;
Molekulární příčinou WA není delece oblasti WAGR, která může souviset s včasným záchytem syndromu WAGR, který se u těchto pacientů vzhledem k jejich nízkému věku ještě nerozvinul (dostupnost podrobných dat z oftalmologického vyšetření.
Podle těchto kritérií byli z další analýzy vyloučeni 3 pacienti bez genových mutací: PAX6 nebo delece chromozomální oblasti 11p13; 3 pacienti bez podrobného popisu klinického obrazu; U 6 pacientů byla zjištěna delece chromozomální oblasti kritické pro rozvoj WAGR syndromu.
Do této analýzy genotypově-fenotypových korelací bylo tedy zahrnuto 98 pacientů s VA ze 73 nepříbuzných rodin (44 sporadických případů a 54 familiárních). Průměrný věk: 16,9±16,8 let (rozmezí: 6 měsíců až 65 let); Většina pacientů ve vzorku (66 osob, 67,3 %) byla vyšetřena před 12. rokem věku. Poměr pohlaví (muž:žena) je 1:1,3. Klinická diagnóza byla stanovena jako výsledek vyšetření pacientů ve Spolkovém státním rozpočtovém vědeckém ústavu „Výzkumné centrum lékařské genetiky“ (52 pacientů) a v pobočce Čeboksary Spolkového státního autonomního ústavu „Lékařsko-vědecký a technický komplex „Mikrochirurgie Oko“ pojmenované po. akad. S. N. Fedorova“ (46 pacientů).
Mezi těmito pacienty byly mutace dříve identifikovány u 6 lidí (74 %) jako výsledek sekvenování exonů a přilehlých oblastí intronů genu PAX75,5. Hlavní část tvoří mutace s posunem otevřeného čtecího rámce a nesmyslné mutace, které dohromady tvoří 2/3 všech identifikovaných intragenních mutací.
U 24 pacientů (24,5 %) byly metodou MLPA identifikovány různé delece oblasti 11p13 [9].
Dále byly vytvořeny skupiny pacientů s genotypy podobnými v molekulárních důsledcích, u kterých byl analyzován fenotyp podle klíčových znaků VA.
Statistická analýza byla provedena pomocí Fisherova exaktního testu.
Výsledky a diskuse
Genetické příčiny VA se dělí do 6 skupin: 4 skupiny drobných genových mutací PAX6: nesmyslné mutace (n=29 pacientů), missense mutace (n=6), mutace sestřihového místa a intronové varianty (n=14), které narušují sestřih, malé inzerce a/nebo delece vedoucí k posunu v otevřeném čtecím rámci (n =19), 2 skupiny velkých chromozomálních přestaveb: ovlivnění oblasti 11p13 bez zohlednění delecí 3′-cis-regulační oblasti genu PAX6 (n=11) a chromozomální delece oblasti 11p13 postihující pouze 3′ cis-regulační oblast (n=13) (tabulka 1).
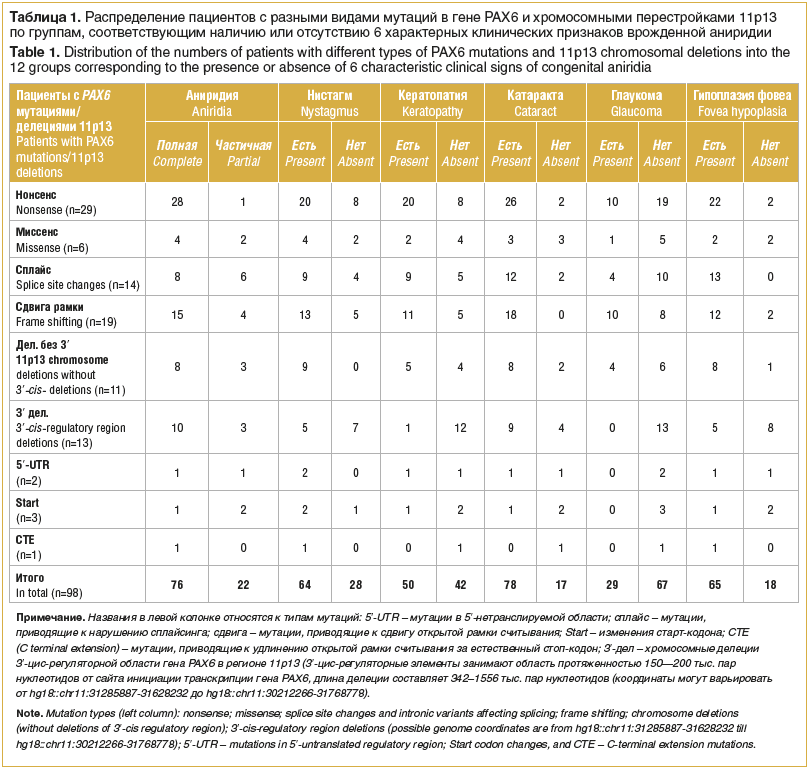
Bylo identifikováno šest charakteristických klinických příznaků ZO: přítomnost kompletní nebo parciální aniridie, nystagmu, keratopatie stadií 6–1, katarakty (bez rozdílu vrozené a komplikované), glaukomu (rovněž bez rozlišení vrozeného a komplikovaného) a hypoplazie centrální retinální fovey.
Pro hledání vztahu mezi charakteristickými fenotypovými rysy často se vyskytujícími u pacientů s VA a detekovanými typy mutací byly sestrojeny a analyzovány kontingenční tabulky 2×2 pomocí Fisherova exaktního testu (tabulka 2).
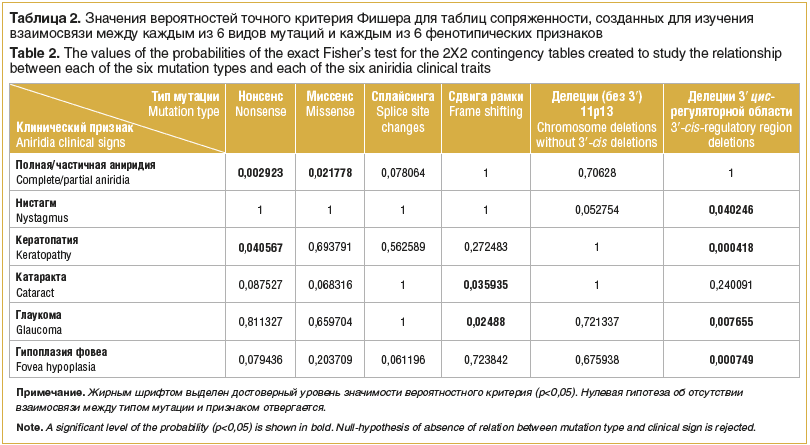
Na základě analýzy bylo identifikováno několik vzorů. U nesmyslných mutací je signifikantně častěji pozorována úplná absence tkáně duhovky a rozvoj keratopatie (p=0,002923 a p=0,040567).
U pacientů s otevřenými mutacemi posunu čtecího rámce je významně vyšší pravděpodobnost rozvoje katarakty a glaukomu (p=0,035935 a p=0,02488, v tomto pořadí).
Částečná aniridie se vyskytuje významně častěji u missense mutací (p=0,021778) než u jiných typů mutací.
Mírnější fenotyp je významně častěji pozorován u pacientů s delecemi 3′-cis-regulační oblasti genu PAX6. Je charakterizována nepřítomností nystagmu, glaukomu, keratopatie a foveální hypoplazie (p=0,040246, p=0,000418, p=0,007655, p=0,00074, v tomto pořadí).
Fenotypy pacientů s chromozomálními delecemi, s výjimkou delecí 3′-cis-regulační oblasti, se neliší od fenotypů pacientů v obecném vzorku a nejsou rozlišeny do samostatné skupiny (p>0,1).
V roce 1998 SK Gupta et al., na základě názoru T. Glaisera et al. o možnosti různého stupně poškození funkcí PAX6 a rozdělení patogenních alel do 3 tříd: amorfní, hypomorfní nebo neomorfní, navrhl a nejprve analyzoval existenci genotypových korelací u VA [12, 13]. Za předpokladu různých rolí pro proteinové izoformy obsahující různé domény SK Gupta et al. navrhl, že některé klinické rysy VA jsou způsobeny poškozením různých domén proteinu PAX6. Podle jejich názoru je katarakta častěji spojena s mutacemi, které ničí doménu PST, a rozvoj poškození sítnice je spojen s mutacemi v párové doméně. Navržená teorie však nebyla potvrzena statisticky spolehlivými metodami, což je pravděpodobně způsobeno malým vzorkem uvažovaných pacientů s VA (n=11).
Naše výsledky jsou částečně v souladu s výše citovanými autory [8, 12, 13]. Teorie T. Glaisera a kol. o třech různých typech mutantních alel PAX6, tedy o různých důsledcích různých typů mutací a původně různých individuálních úrovních exprese PAX6 pod vlivem genetického pozadí dokonale vysvětluje klinickou heterogenitu VA, odhalenou také v naší studii [12]. I. Tzulaki et al., kteří nenašli žádné statisticky významné korelace mezi fenotypem a genotypem, vysvětlují vyhlazování rozdílů v důsledcích nonsensových mutací a mutací s posunem otevřeného čtecího rámce různých lokalizací existencí univerzálního mechanismu degradace mRNA. s předčasným stop kodonem (NMD), bez ohledu na to, kde je lokalizována mutace vedoucí k rozvoji PTS [8]. Nicméně SK Gupta a spol. měly částečně pravdu v tom, že umístění mutace může ovlivnit typ poškození funkce PAX6.
K testování hypotézy SK Gupta et al. Byla provedena dodatečná analýza, pro kterou byly mutace identifikované v naší studii rozděleny do 6 skupin na základě principu jejich lokalizace. Byla analyzována možná závislost každého ze 6 charakteristických klinických příznaků VA na lokalizaci mutací v různých doménách proteinu PAX6 (tabulka 3).
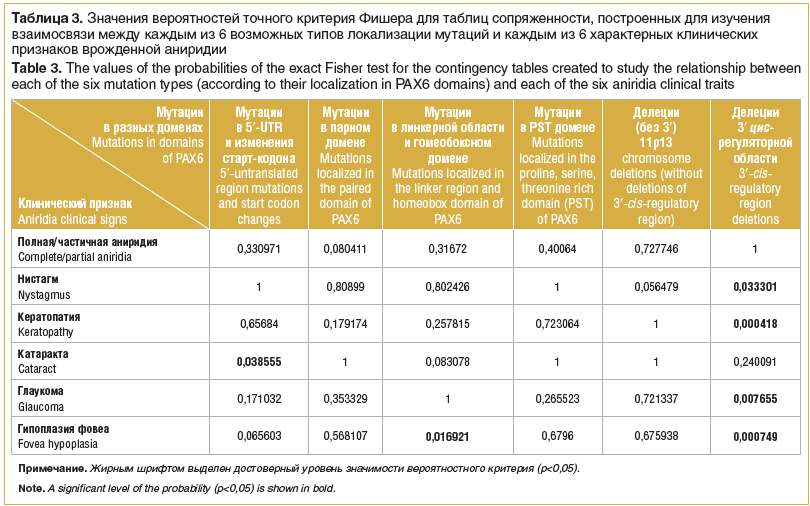
Tato analýza ukázala, že na rozdíl od údajů SK Gupta et al. [12], pouze mutace v oblasti linkeru a homeodomény jsou významně častěji spojeny s foveální hypoplazií (p=0,016921). Spolehlivá závislost byla získána také pro rys vývoje katarakty a malou skupinu mutací v 5′-UTR a změny ve start kodonu.
V naší práci jsme rozdělili mutace nikoli podle principu jejich lokalizace v určitých proteinových doménách PAX6, ale podle kategorií, které pravděpodobně odrážejí typy poškození funkce. Plně jsme tedy dodrželi princip teorie T. Glaisera a kol. na korespondenci klinického obrazu s typem mutantní alely: hypomorfní v různé míře nebo zcela amorfní [13]. Hypomorfní alely jsou pravděpodobně produkovány missense mutacemi a delecemi 3′ cis-regulační oblasti genu PAX6bez ovlivnění jeho kódovací sekvence. Odpovídají mírnějším fenotypům. Amorfní alely se ztrátou funkce jsou produkovány nesmyslnými a posunovými mutacemi, zejména pokud tyto mutace poškozují N-konec proteinu (párové a homeodomény), stejně jako chromozomální delece 11p13, které zahrnují kódující sekvenci genu PAX6.
Závěr
Vzhledem k vysoké genetické heterogenitě a klinickému polymorfismu nebyly genotypové korelace u VA dosud stanoveny. Především je to dáno potřebou dostatečného počtu pacientů v analyzovaném vzorku a dostupností podrobného popisu klinického obrazu ZO u těchto pacientů. Na vzorku 98 pacientů bylo možné poprvé studovat a identifikovat genotypové korelace ve skupině pacientů s VA. Byly analyzovány možné vztahy mezi každým ze 6 typů mutací v genu PAX6 (nesmysl, posun otevřeného čtecího rámce, missense, sestřih nebo delece 11p13 ovlivňující pouze regulační oblast nebo kódující sekvenci genu PAX6) a každý z hlavních charakteristických fenotypových rysů VA.
Jako výsledek studie byly poprvé stanoveny následující vzorce. U nonsensových mutací je výrazně častěji pozorována úplná absence duhovkové tkáně a rozvoj keratopatie, u otevřených čtecích mutací s posunem čtecího rámce se výrazně častěji rozvíjí katarakta a glaukom, proto jsou fenotypy spojené s mutacemi vedoucími ke ztrátě funkce charakterizovány závažnějším klinický průběh, který možná v důsledku významného poškození funkce genu v důsledku úplné ztráty jedné alely. Nesynonymní substituce jsou spojeny se stejným VA fenotypem jako v obecném vzorku, ale parciální aniridie se vyskytuje významně častěji u missense mutací než u jiných typů mutací. Fenotypy pacientů s chromozomálními delecemi, s výjimkou delecí 3′ cis-regulační oblasti, se neliší od fenotypů pacientů s intragenní PAX6 mutace. U pacientů s delecemi je pozorován mírnější fenotyp napříč několika klinickými rysy
3′ cis-regulační oblast genu PAX6.
Identifikace klinických příznaků VA v závislosti na detekovaných mutacích je nejen klinicky, ale i významně vědecky zajímavá, protože ukazuje na jeden z mechanismů regulace genových funkcí, který skutečně funguje v lidském těle. PAX6.