Symptomy a léčba Konovalov-Wilsonovy choroby
Patogenní varianta c. 2532dela v genu ATP7B, patogenní varianta c.1340_1343del4 v genu ATP7B, patogenní varianta c.1770insT v genu ATP7B, patogenní varianta c.2304insC v genu ATP7B, patogenní varianta H1069QE1064Q ve screeningu patogenní varianty genu ATP7. 3026_3028delTCA, 3029insT, 3031insC v genu ATP7B, patogenní varianta c.3402delC v genu ATP7B, screening patogenních variant c.3627_3630del4, 3649_3654del6 v genu c.7delC, screening patogenní varianty genu ATP3942B 3947delAT, 7delG v genu ATPXNUMXB
Metoda měření:
PCR a fragmentová analýza
Jednotky:
Kvalitativní výzkum
Wilsonova choroba (WD) je vzácná autozomálně recesivně dědičná porucha metabolismu mědi. Tento stav je charakterizován nadměrným ukládáním mědi v játrech, mozku a dalších tkáních. Hlavním patogenetickým mechanismem onemocnění je nadměrné vstřebávání mědi z tenkého střeva a snížené vylučování mědi játry.
Asi 50%-75% mědi je absorbováno ve střevě a poté transportováno do hepatocytů. Tato cesta není ovlivněna u Wilsonovy choroby. Jakmile se měď dostane do hepatocytů, je začleněna do enzymů obsahujících měď a proteinů vázajících měď (CBP), včetně ceruloplasminu a sérové ferroxidázy.
Normálně se přebytek mědi váže na apo-metallothionein za vzniku komplexů nebo je vylučován žlučí. Rovnováha mědi v těle je udržována spíše regulací vylučování než absorpce a vylučování mědi probíhá převážně (přibližně 95 %) hepatobiliárním systémem.
U Wilson-Konovalovovy choroby jsou narušeny procesy inkorporace mědi do ceruloplasminu a vylučování přebytečné mědi do žluči. Transport mědi ATPázou typu P přenášející měď je defektní v důsledku dědičného defektu v genu ATP7B. Podle výsledků genetických studií jsou hlavní lokusy, ve kterých se poruchy vyskytují, lokalizovány v 13q14-q21.
Mnoho defektů genu ATP7B jsou malé delece, inzerce nebo patogenní varianty. Většina pacientů nese různé aberace na každém ze 2 chromozomů. Bylo identifikováno více než 40 různých patogenních variant, z nichž nejčastější je substituce histidinu za glutamin v pozici 1069 (H1069Q). Bylo také zjištěno, že patogenní varianta H1069Q je spojena s pozdními neurologickými projevy.
Nadbytek mědi v důsledku Wilsonovy choroby podporuje tvorbu volných radikálů, což vede k oxidaci lipidů a bílkovin. Již v časných stadiích hepatocelulárního poškození lze detekovat ultrastrukturální abnormality v endoplazmatickém retikulu, mitochondriích, peroxisomech a jádrech. Zpočátku se nadbytek mědi hromadí v játrech, což vede k poškození hepatocytů. Později, když dochází k nadměrnému ukládání mědi v játrech, její koncentrace v cirkulující krvi se zvyšuje a následně dochází k jejímu ukládání v jiných orgánech a systémech.
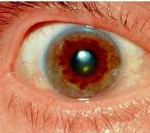
Wilson-Konovalovova choroba — je vzácná genetická porucha, která začíná dysfunkcí jater. Obvykle se léze vyvíjí do šesti let, ale klinické projevy jsou častěji detekovány v dospívání nebo ve věku 20 let. Příznaky onemocnění zahrnují: žloutenku (žluté zbarvení kůže, sliznic a oční bělmy), otoky nohou a zvětšení objemu břicha (ascites) v důsledku porušení onkotického tlaku plazmy , tvorba abnormálních cévních anastomóz ve formě křečových žil jícnu, které mohou způsobit krvácení, stejně jako sklon k tvorbě modřin a prodloužení doby krvácení. Někteří pacienti s Wilsonovou chorobou mohou mít po mnoho let pouze drobné abnormality v jaterních testech a žádné další příznaky. Charakteristický patognomický příznak ukládání mědi ve formě hnědého okraje na rohovce oka (Kayser-Fleischerovy kroužky) se obvykle tvoří ve věku 40 let a lze jej zřídka detekovat u dětí. Kayser-Fleischnerův prstenec je nekonzistentní nález a je pozorován u méně než 50 % pacientů.
U některých pacientů může dojít k selhání jater. V tomto případě lze pozorovat rychlý nárůst příznaků jaterního selhání s rozvojem hemolytické anémie a deprese vědomí až po jaterní encefalopatii a kóma.
U některých pacientů se jaterní dysfunkce nemusí projevit až po mnoha letech a pak se do popředí klinického obrazu dostávají neurologické poruchy. Mezi běžné neurologické příznaky Wilsonovy choroby, které se mohou časem vyvinout a progredovat, patří třes, mimovolní pohyby, potíže s polykáním (dysfagie), problémy s řečí a artikulací (dysartrie), nedostatek koordinace, spasticita a svalová ztuhlost. Téměř všichni pacienti s neurologickými příznaky mají Kayser-Fleischerovy kroužky, které lze detekovat při oftalmologickém vyšetření.
Psychiatrické projevy Wilsonovy choroby se mohou u jednotlivých pacientů značně lišit. Tyto příznaky jsou často zaměňovány s jinými nemocemi, od deprese po schizofrenii, a jsou často mylně diagnostikovány jako zneužívání návykových látek. Pacienti často pociťují změny osobnosti a poruchy chování. U většiny pacientů s psychiatrickými příznaky se současně rozvinou i neurologické příznaky, případně se rozvinou do tří let.
U mladých žen může dojít k opožděnému nástupu menstruace nebo může menstruace přestat před zahájením léčby. To je způsobeno obecnou změnou metabolismu hormonů v důsledku dysfunkce jater způsobené Wilson-Konovalovovou chorobou. Mezi pacientkami se také vyskytují další poruchy menstruačního cyklu, včetně amenorey, stejně jako potratů a neplodnosti.
Mezi další projevy Wilsonovy choroby patří urolitiáza a poškození ledvinových tubulů, časná artritida a další kostní a kloubní léze, včetně osteoporózy a osteofytózy velkých kloubů. Dochází také ke zúžení kloubních prostor kloubů páteře a končetin.
Diagnostika BV je založena na kombinovaném posouzení pacientovy anamnézy, klinického nálezu a specifických testů. Taková vyšetření mohou zahrnovat oftalmologické vyšetření očí štěrbinovou lampou k detekci Kayser-Fleischerových prstenců, hladiny ceruloplasminu v séru (snížené) a vylučování mědi močí (zvýšené). Ve vzácných případech lze doporučit jaterní biopsii s následným morfologickým vyšetřením depozit mědi v orgánovém parenchymu.
Nejdůležitější fází diagnostiky je molekulárně genetické vyšetření. Protože v literatuře bylo popsáno více než 300 patogenních variant genu ATP7B, testem volby je sekvenování lokusů odpovědných za vývoj patologie. Vzhledem k tomu, že aberace H1069Q se u evropské populace vyskytuje u 30–60 % případů onemocnění, je vhodné ji stanovit pomocí alelově specifické PCR v kombinaci se sekvenační metodou. Pokud jsou zjištěny výše uvedené genetické poruchy, může být pacientům doporučeno genetické poradenství jejich přímých příbuzných pro asymptomatické přenášení aberací spojených s Wilsonovou-Konovalovovou chorobou.
Je nesmírně důležité diagnostikovat BV v raných fázích vývoje. Právě včasná diagnostika a včasná léčba pomáhají vyhnout se chronickému selhání jater a neurologické dysfunkci.
- Přítomnost homozygotní aberace v genu ATP7B nebo složená heterozygotnost (přenašeč 2 různých heterozygotních aberací) potvrzuje diagnózu Wilson-Konovalovovy choroby.
- Nošení jedné heterozygotní aberace v přítomnosti charakteristického klinického obrazu onemocnění významně zvyšuje pravděpodobnost diagnózy Wilson-Konovalovovy choroby. Takoví pacienti také nemusí mít žádné specifické příznaky onemocnění a jsou přenašeči schopnými přenést genetický defekt na své potomky.
- Absence běžných aberací v genu ATP7B významně snižuje pravděpodobnost diagnózy Wilson-Konovalovovy choroby, ale nevylučuje ji. Za přítomnosti charakteristických klinických příznaků může být takovému pacientovi doporučeno podstoupit podrobnější studium struktury genu ATP7B.
Wilsonova choroba, hepatolentikulární degenerace, hepatocerebrální dystrofie, dědičná intoxikace mědí, Westphal-Wilsonova choroba, Wilsonova choroba, hepatolentikulární degenerace, ATP7B, hepatitida, hepatóza,
Wilsonova nemoc – dědičné onemocnění přenášené autozomálně recesivním způsobem. Dochází k němu v důsledku mutací v genu ATP7B, který kóduje jaterní protein ATPázy přenášející měď. Charakteristickým příznakem Wilsonovy choroby je hromadění mědi v různých orgánech a tkáních, většinou v játrech a bazálních gangliích. Wilsonova choroba se může vyskytovat v abdominální, rigidně-arytmicko-hyperkinetické, tremulosní nebo extrapyramidově-kortikální formě. Diagnostika Wilsonovy choroby zahrnuje oftalmologické vyšetření, biochemické testy moči a krve a MRI nebo CT vyšetření mozku. Základem patogenetické terapie jsou thiolové léky, které lze užívat několik let i doživotně.
ICD-10
E83.0 Poruchy metabolismu mědi
- Příčiny
- Klasifikace
- Příznaky
- diagnostika
- Léčba Wilsonovy choroby
- Prognóza a prevence
- Ceny za ošetření
Přehled
Wilsonova nemoc – dědičné onemocnění přenášené autozomálně recesivním způsobem. Dochází k němu v důsledku mutací v genu ATP7B, který kóduje jaterní protein ATPázy přenášející měď. Charakteristickým příznakem Wilsonovy choroby je hromadění mědi v různých orgánech a tkáních, většinou v játrech a bazálních gangliích. Objevitelem nemoci je A.K. Wilson, který nemoc popsal v roce 1912, v domácí medicíně – N.A. Konovalov. Patogeneze Wilsonovy choroby byla identifikována v roce 1993. Pojem „Wilsonova choroba“ také odpovídá: Wilson-Konovalovově chorobě, Westphal-Wilson-Konovalovově chorobě, hepatocerebrální dystrofii, hepatolentikulární dystrofii, progresivní lentikulární degeneraci.
Příčiny
Gen ATP7B je mapován na dlouhém rameni chromozomu 13 (13q14.3-q21.1). Lidské tělo obsahuje asi 50-100 mg mědi. Denní potřeba mědi pro člověka je 1-2 mg. 95 % mědi absorbované ve střevě je transportováno ve formě komplexu s ceruloplasminem (jeden ze sérových globulinů syntetizovaných játry) a pouze 5 % ve formě komplexu s albuminem. Kromě toho je iont mědi součástí nejdůležitějších metabolických enzymů (lysyloxidáza, superoxiddismutáza, cytochrom C oxidáza atd.). U Wilsonovy choroby jsou narušeny dva procesy metabolismu mědi v játrech – biosyntéza hlavního proteinu vázajícího měď (ceruloplasminu) a vylučování mědi žlučí, což má za následek zvýšení hladiny nevázané mědi v krvi. Zvyšuje se koncentrace mědi v různých orgánech (nejčastěji v játrech, ledvinách, rohovce a mozku), což vede k jejich toxickému poškození.
Klasifikace
Podle klasifikace N.V. Konovalov rozlišuje pět forem Wilsonovy choroby:
- břišní
- rigidní arytmická hyperkinetika
- chvění-tuhý
- chvění
- extrapyramidově-kortikální
Příznaky
Wilsonova choroba je charakterizována klinickým polymorfismem. První projevy onemocnění se mohou objevit v dětství, dospívání, dospělosti a mnohem méně často v dospělosti. Ve 40-50% případů se Wilsonova choroba projevuje poškozením jater, ve zbytku – duševními a neurologickými poruchami. Se zapojením nervového systému do patologického procesu je detekován Kayser-Fleischerův prstenec.
Tvar břicha rozvíjí se především před 40. rokem věku. Charakteristickým příznakem je těžké poškození jater, jako je cirhóza jater, chronická hepatitida, fulminantní hepatitida.
Rigidně-arytmická hyperkinetická forma se projevuje v dětství. Počáteční projevy jsou svalová ztuhlost, amimie, nezřetelná řeč, potíže s prováděním malých pohybů a mírný pokles inteligence. Tato forma onemocnění je charakterizována progresivním průběhem s epizodami exacerbace a remise.
třesoucí se forma vyskytuje se ve věku 10 až 30 let. Převládajícím příznakem je třes. Kromě toho mohou být pozorovány bradykineze, bradyllie, těžký psychoorganický syndrom a epileptické záchvaty.
Extrapyramidově-kortikální forma je velmi vzácný. Jeho začátek je podobný začátku kterékoli z výše uvedených forem. Je charakterizována epileptickými záchvaty, extrapyramidovými a pyramidovými poruchami a těžkým intelektuálním deficitem.
diagnostika
Oftalmologické vyšetření pomocí štěrbinové lampy odhalí Kayser-Fleischerův prstenec. Biochemické studie moči odhalují zvýšené vylučování mědi v denní moči, stejně jako snížení koncentrace ceruloplasminu v krvi. Pomocí zobrazovacích metod (CT a MRI mozku) se zjišťuje atrofie mozkové a cerebelární hemisféry a také bazálních ganglií.
Při diagnostice Wilsonovy choroby ji musí neurolog odlišit od Parkinsonismu, hepatocerebrálního syndromu a Hellervorden-Spatzovy choroby. Hlavním diferenciálně diagnostickým znakem těchto onemocnění je absence Kayser-Fleischerova prstence a poruchy metabolismu mědi charakteristické pro Wilsonovu chorobu. K potvrzení Wilsonovy choroby se provádí genová diagnostika.
Léčba Wilsonovy choroby
Základem patogenetické léčby je předepisování thiolových léků, především D-penicilaminu nebo unitiolu. Hlavní výhodou cuprenilu je nízká toxicita a možnost dlouhodobého užívání bez vedlejších účinků. Předepisuje se 0,15 g (1 kapsle) denně (pouze po jídle), poté se během 2,5-3 měsíců dávka zvyšuje na 6-10 kapslí/den (optimální dávka). Léčba D-penicilaminem se provádí roky a dokonce i celoživotně s krátkými přestávkami (2-3 týdny) v případě nežádoucích účinků (trombocytopenie, leukopenie, exacerbace žaludečního vředu atd.).
Unithiol se předepisuje v případě intolerance (špatné snášenlivosti) D-penicilaminu. Délka jednoho léčebného cyklu je 1 měsíc, poté je léčba přerušena na 2,5-3 měsíce. Ve většině případů dochází ke zlepšení celkového stavu pacienta a také k ústupu neurologických symptomů (ztuhlost, hyperkineze). V případě dominance hyperkineze se doporučuje předepisovat malé cykly antipsychotik a pro rigiditu – levodopa, karbidopa, trihexyfenidyl.
V případech těžké Wilsonovy choroby, pokud je konzervativní léčba v zahraničí neúčinná, se používá transplantace jater. Pokud je výsledek operace pozitivní, stav pacienta se zlepšuje a metabolismus mědi v těle je obnoven. Další léčbou pacienta je imunosupresivní léčba. V Rusku se dnes do klinické praxe postupně zavádí metoda biohemoperfuze s izolovanými živými buňkami sleziny a jater (tzv. přístroj „pomocná játra“). Nemedikamentózní léčba spočívá v předepsání diety (tabulka č. 5) s cílem vyloučit potraviny bohaté na měď (káva, čokoláda, luštěniny, ořechy atd.).
Prognóza a prevence
V případě včasné diagnózy Wilsonovy choroby a adekvátní terapie snižující hladinu mědi je možná normalizace celkového stavu pacienta a metabolismu mědi v těle. Kontinuální příjem thiolových léků podle režimu předepsaného lékařským specialistou umožňuje pacientovi udržovat profesionální a společenskou aktivitu.
Aby se zabránilo relapsům Wilsonovy choroby, doporučuje se několikrát ročně provést laboratorní testy krve a moči pacienta. Je třeba sledovat tyto ukazatele: koncentraci mědi, ceruloplasminu a zinku. Kromě toho se doporučuje provést biochemický krevní test, obecný krevní test a také pravidelné konzultace s terapeutem a neurologem.