Kraniosynostóza: 4 příčiny rozvoje, doprovodný klinický obraz a 4 diagnostické metody
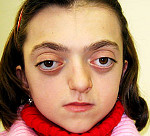
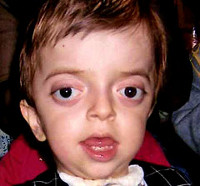
Crusonův syndrom – vzácné genetické onemocnění provázené progresivními deformacemi obličejové a mozkové části lebky a kraniosynostózou s rozvojem přidružených poruch. Mezi příznaky tohoto stavu patří změny tvaru hlavy (brachycefalie, skafocefalie, trigonocefalie), hákovitý nos, hypoplazie střední třetiny obličeje a poruchy zraku a sluchu. Diagnóza Crouzonova syndromu je založena na vnějších projevech onemocnění, radiologických datech a molekulárně genetické analýze. Pro tuto patologii neexistuje žádná specifická léčba a používají se paliativní a symptomatická opatření, včetně chirurgických.
- Příčiny Crouzonova syndromu
- Příznaky Crouzonova syndromu
- Diagnóza Crouzonova syndromu
- Léčba Crouzonova syndromu
- Prognóza a prevence Crouzonova syndromu
Přehled
Crouzonův syndrom (kraniofasciální dysostóza typu 1) je genetické onemocnění charakterizované narušením osifikačních procesů a rozvojem kosterních prvků obličejové a kraniální lebky. Tento stav byl poprvé popsán v roce 1912 francouzským pediatrem O. Crouzonem a od té doby nese syndrom jeho jméno. Mechanismus dědičnosti Crouzonova syndromu je autozomálně dominantní, ale onemocnění je často způsobeno spontánními mutacemi. Patologie je poměrně vzácná – přibližně 1,6 případů na 100 000 novorozenců, přičemž tento syndrom způsobuje téměř 5% všech vývojových vad doprovázených kraniální dysostózou. Po dlouhou dobu se věřilo, že tento stav má dvě varianty – normální a doprovázený kožními poruchami (hyperkeratóza, akantóza), ale s ohledem na moderní údaje odborníci v oblasti genetiky zjistili, že Crouzonův syndrom s acanthosis nigricans (CAN ) je samostatné dědičné onemocnění. Zároveň je patogeneze jeho vývoje podobná klasické formě onemocnění, což vysvětluje významnou podobnost příznaků. Tento stav pravděpodobně postihne chlapce i dívky.

Příčiny Crouzonova syndromu
Klasická verze Crouzonova syndromu je způsobena mutacemi v genu FGFR2, lokalizovaném na 10. chromozomu – kóduje aminokyselinovou sekvenci receptoru pro fibroblastový růstový faktor 2. Tento gen je významné velikosti a má velký počet exonů, které snižuje jeho stabilitu – často se v něm vyvíjejí defekty, které vedou k četným genetickým onemocněním postihujícím především kosterní prvky. Mutace FGFR2 tak mohou být vedle Crouzonova syndromu příčinou Apertových, Saethre-Chotzenových, Beer-Stevensonových syndromů, Pfeifferova syndromu a mnoha dalších patologií. Genetické studie prokázaly, že kraniofaciální dysostózu typu 1 může způsobit více než 35 mutací výše uvedeného genu, lokalizovaných především v oblasti exonů 7 a 9.
Téměř všechny defekty v genu FGFR2 jsou missense mutace, to znamená, že vyvolávají změnu ve struktuře kódovaného proteinu. Konformační změny v receptoru fibroblastového růstového faktoru 2 narušují mezibuněčné interakce v pojivových tkáních lebky, především kosti a chrupavce. To vede nejprve k akumulaci fibroblastů v oblasti interoseálních stehů a poté k aktivaci osifikačních procesů, což je příčinou hlavní manifestace Crouzonova syndromu – kraniální synostózy. Někteří badatelé se domnívají, že tyto genetické vady ovlivňují i embryonální vývoj struktur prvního větevního oblouku – mezi ně patří čelisti a do jisté míry i prvky střední třetiny obličeje. To vysvětluje hypoplazii čelistí, zejména dolní, u Crouzonova syndromu.
Příčiny Crouzonova syndromu s acanthosis nigricans jsou poněkud odlišné – je způsoben mutacemi v genu FGFR3, lokalizovaným na chromozomu 4. Produktem jeho exprese je také receptor pro fibroblastový růstový faktor, pouze typu 3 (na rozdíl od typu 2, který je produktem genu FGFR2). Bylo zjištěno, že pouze jedna mutace tohoto genu způsobuje Crouzonův syndrom s charakteristickými kožními projevy – Ala391Glu. Toto je také missense mutace, která mění strukturu receptorového proteinu. Patogeneze onemocnění se prakticky neliší od klasické varianty. Změny v obličeji a lebce u Crouzonova syndromu s acanthosis nigricans jsou podobné předchozímu typu, ale jsou doprovázeny hyperkeratózou různých oblastí kůže a akantózou a často jsou pozorovány četné moly.
Příznaky Crouzonova syndromu
Projevy Crouzonova syndromu lze zaznamenat již při narození dítěte, nejvýrazněji se však projevují během prvních 3-4 let života. Nejcharakterističtějším příznakem onemocnění je kraniosynostóza, která se může vyvinout na koronálním nebo sagitálním (mnohem méně často) stehu, pevně spojující kosti a zastavující normální růst hlavy. Ihned po narození mohou být první známky synostózy vymazány, ale hypertelorismus, prognatismus dolní čelisti, změna tvaru nosu jako „papouší zobák“, mírný exoftalmus v důsledku zmenšení očních důlků a vždy je pozorována nízká poloha zevního zvukovodu. Někdy je Crouzonův syndrom doprovázen syndaktylií prstů, v tomto případě je nutné provést diferenciální diagnostiku s Apertovým syndromem. U některých pacientů se rozvine choanální atrézie, která ztěžuje dýchání, a také hydrocefalus, který dále komplikuje průběh onemocnění v důsledku prudkého zvýšení intrakraniálního tlaku.
Charakteristickým znakem Crouzonova syndromu je nevyhnutelná progrese onemocnění, zejména ve vztahu k tvaru lebky. V důsledku tvorby silné synostózy a pokračujícího růstu mozku se mění tvar hlavy, dochází k brachycefalii nebo „věžové lebce“ podle toho, který steh srostl. U Crouzonova syndromu se exostózy mohou tvořit také v oblasti srostlých lebečních kostí. Tato deformace vede i k poškození zrakových orgánů – nejprve vzniká divergentní strabismus, poté silně progreduje exoftalmus, až oční bulvy vypadnou z očnice. Crouzonův syndrom je často doprovázen poruchami sluchu v důsledku porušení struktury pyramidy spánkové kosti – její dutiny jsou zmenšeny, některé z nich mohou chybět, což často vede k úplné hluchotě. Změny jsou pozorovány i na nervovém systému, jsou zjišťovány přibývající známky mentální retardace (při absenci paliativních opatření), příznaky zvýšeného intrakraniálního tlaku (bolesti hlavy, zvracení) a záchvaty.
Crouzonův syndrom s acanthosis nigricans je charakterizován podobnými změnami na obličeji a lebce. Někteří vědci zároveň poznamenávají, že tato forma onemocnění je obecně závažnější a vyznačuje se zvýšenou frekvencí komplikací. Choanální atrézie, která se u klasické variety kraniofaciální dysostózy 1. typu vyvine zcela vzácně, je tedy zaznamenána u téměř poloviny pacientů s Crouzonovým syndromem s acanthosis nigricans. Dále se u pacientů objevují závažné kožní poruchy – hyperkeratóza (proliferace bradavic, kožní hypertrofie), zvýšená pigmentace. Hlavní lokalizací kožních projevů u Crouzonova syndromu s acanthosis nigricans jsou oblasti kolenních a loketních záhybů, krku, břicha, nasolabiálních rýh a oblast kolem očí. Toto onemocnění je také charakterizováno přítomností velkého počtu névů (mol), často se vyvíjejí hypertrofické, slabě pigmentované jizvy a jizvy.
Diagnóza Crouzonova syndromu
Crouzonův syndrom lze detekovat v prenatálním stadiu, bezprostředně po narození nebo v prvních letech života pacienta. K tomuto účelu se používají rentgenové metody, obecné vyšetření a molekulárně genetická analýza. Podpůrnou roli v diagnostice Crouzonova syndromu hrají takové metody, jako je oftalmologické vyšetření, vyšetření sluchu a posouzení intelektuálního a duševního vývoje. Při vyšetření malých dětí se zjišťují nízko nasazené uši, hypoplazie střední třetiny obličeje a exoftalmus. U starších pacientů s Crouzonovým syndromem jsou tyto příznaky doprovázeny divergentním šilháním, ztrátou sluchu až úplnou hluchotou a změnami tvaru lebky. Rentgen lebky odhaluje synostózu v oblasti koronálních, sagitálních nebo lambdoidních stehů a lze detekovat zploštělý tvar orbit;
Tomografie spánkové kostní pyramidy u Crouzonova syndromu odhaluje poruchu tvorby zevního zvukovodu (atrézie nebo stenóza) a někdy je pozorována absence bubínkové dutiny. Sella turcica je poněkud rozšířena a mohou se tvořit další malé paranazální dutiny. Molekulárně genetická diagnostika Crouzonova syndromu je prováděna genetikem a u klasické formy onemocnění je omezena na automatické sekvenování exonů 7 a 9 genu FGFR2 za účelem identifikace mutací. Při výskytu kožních projevů (hyperkeratóza, bradavice, mnohočetná znaménka) má smysl pátrat po mutaci Ala391Glu v genu FGFR3. U obou forem Crouzonova syndromu je možná prenatální genetická diagnostika, ale ultrazvukové techniky jsou obecně neúčinné.
Léčba Crouzonova syndromu
V současné době neexistuje žádná specifická léčba Crouzonova syndromu, používají se pouze paliativní opatření. Patří mezi ně chirurgické zákroky k remodelaci tvaru lebky a odstranění synostóz – takové zákroky je třeba zahájit co nejdříve a poté je s růstem hlavy provést ještě několikrát. Tím se snižuje hladina intrakraniálního tlaku, což má pozitivní vliv na duševní vývoj pacientů s Crouzonovým syndromem a snižuje pravděpodobnost neurologických poruch. Také pomocí chirurgických technik se vytváří umělá blefarofimóza, aby se snížil stupeň exoftalmu a zabránilo se dislokaci oční bulvy. V případě choanální atrézie se chirurgicky rozšiřují, aby se usnadnilo dýchání. Jsou popsány techniky radikálních komplexních operací zaměřených na odstranění většiny obličejových poruch u Crouzonova syndromu. V případě vývoje kožních změn se doporučuje vnější použití přípravků na bázi retinoidů ke snížení jejich závažnosti, někdy jsou předepsány kortikosteroidy.
Prognóza a prevence Crouzonova syndromu
Prognóza Crouzonova syndromu je obecně nejistá, řada odborníků ji hodnotí jako nepříznivou. Je to dáno tím, že i při všech symptomatických a paliativních opatřeních se u pacientů stále objevuje narůstající divergentní strabismus, téměř vždy se časem rozvine hluchota a s věkem se zvýrazňuje hypoplazie střední třetiny obličeje. Mnoho pacientů se však při vhodné léčbě a péči může dožít vysokého věku. Kvůli těžkému zrakovému a sluchovému postižení se pacienti téměř vždy mohou stát příčinou invalidity; Prevence Crouzonova syndromu nebyla vyvinuta; možné je pouze prenatální stanovení patologie pomocí molekulárně genetických metod.