Potřeba léčby Huntingtonovy chorey. Huntingtonova chorea (Huntingtonova choroba) – popis syndromu, diagnostika a možnosti léčby
Huntingtonova chorea (Huntingtonova choroba) je závažné dědičné neurodegenerativní onemocnění charakterizované progresivní destrukcí mozkových struktur.
Tato patologie je spojena s mutací v genu odpovědném za produkci proteinu huntingtin. Změněný gen vede k akumulaci abnormálního proteinu v nervových buňkách a rozvoji jejich dysfunkce. Postupem času jsou ovlivněny oblasti mozku, které regulují koordinaci, mentální funkce a autonomní procesy.
Hlavním projevem onemocnění je chorea – mimovolní záškuby hyperkinetické pohyby. Kromě toho se rozvíjejí kognitivní poruchy a poruchy chování, psychózy a deprese. Progresivní atrofie mozku nakonec vede k úplné invaliditě a předčasné smrti pacientů.
Charakteristiky této nemoci
Huntingtonova chorea má řadu charakteristických rysů. Znalost těchto charakteristických znaků je nesmírně důležitá jak pro včasnou diagnostiku, tak pro pochopení patogenetických mechanismů rozvoje onemocnění, které je nezbytné pro vývoj účinných léčebných režimů.
- Dědičnost. Onemocnění se dědí autozomálně dominantním způsobem, což znamená, že existuje 50% pravděpodobnost přenosu mutovaného genu na potomky od postiženého rodiče.
- Plný penetrant. Pokud člověk zdědí defektní gen, pak s přibývajícím věkem se u něj nevyhnutelně rozvinou příznaky Huntingtonovy choroby.
- Progresivní kurz. Příznaky mají tendenci se v průběhu času neustále zvyšovat. Doba trvání onemocnění po manifestaci je zpravidla 15-20 let.
- Odložený debut. První projevy Huntingtonovy choroby se obvykle objevují mezi 35. a 50. rokem života.
- Multisystémové poškození. Patologický proces postihuje nejen pohybovou sféru, ale i vyšší psychické funkce a autonomní regulaci.
Vezmeme-li tyto rysy v úvahu při vývoji léčebného režimu pro Huntingtonovu choreu, je možné zpomalit progresi symptomů a zlepšit kvalitu života pacientů.
Příčiny onemocnění
Huntingtonova chorea je genetická porucha. Příčinou je mutace jednoho genu, HTT, umístěného na krátkém raménku 4. chromozomu. Tento gen kóduje protein zvaný huntingtin, který hraje důležitou roli v nervových přenosových procesech v mozku. Právě změny v něm stojí za vznikem onemocnění.
U pacientů s Huntingtonovou choreou má DNA tohoto genu expanzi opakujícího se tripletu CAG. Kritická délka těchto repetic je 36 nebo více, což činí gen mutantem. Protein na jeho základě získává abnormální strukturu a začíná se hromadit v neuronech, včetně bazálních ganglií a mozkové kůry. To vede k degenerativním procesům a atrofii nervové tkáně.
Včasná detekce nosičství takového mutantního genu je extrémně důležitá pro dynamické sledování a včasné zahájení léčby v případě prvních příznaků Huntingtonovy chorey.

„Nemoc sama od sebe nezmizí, převezměte zodpovědnost za své zdraví. Existují nějaké příznaky? Přihlaste se k nám na konzultaci.“
Vedoucí lékařka T. L. Kharaburova
Přihlaste se ke konzultaci na telefonním čísle 8 (831) 266-03-06 nebo zanechte žádost. Přihlásit se
Fáze a formy Huntingtonovy choroby
Existuje 5 fází onemocnění:
- Presymptomatické (počáteční) stadium. V této fázi ještě nejsou patrné klinické projevy, lze však pozorovat nespecifické změny osobnosti, ztrátu paměti a pozornosti.
- Rané (počáteční) stadium trvá 2-10 let. Objevují se první poruchy motorické (choreiformní hyperkineze), kognitivní, behaviorální a psychické poruchy.
- Pokročilé (střední) stadium může trvat od 2 do 15 let. Poruchy hybnosti progredují, zvyšuje se pokles intelektu a paměti a rozvíjejí se behaviorální a psychotické abnormality.
- Pozdní fáze trvá od 2 do 10 let. Choreiformní hyperkineze, demence, poruchy řeči a polykání jsou ostře vyjádřeny. Postižení přibývá.
- Terminální fáze je poslední fáze, která trvá od 1 roku do 5 let. Charakterizováno celkovým psychoorganickým úpadkem, neschopností postarat se o sebe a vegetativními poruchami.
Tato periodizace je důležitá pro pochopení dynamiky onemocnění a volbu adekvátní taktiky léčby v každé fázi.
Kromě stagingu onemocnění existuje několik klinických forem Huntingtonovy chorey, které se liší v závislosti na věku manifestace a závažnosti příznaků.
Fáze odrážejí dynamiku progrese onemocnění a formy charakterizují rysy jejího průběhu v každém konkrétním případě.
- Juvenilní forma – nástup onemocnění před 20. rokem života. Vyznačuje se závažnějším a rychle progredujícím průběhem.
- Manifestní (klasická) forma – debut mezi 30. a 50. rokem. Nejčastější varianta onemocnění.
- Pozdní forma – nástup onemocnění po 50 letech. Vyznačuje se poněkud pomalejším rozvojem příznaků oproti klasické variantě.
- Abortivní forma – pokud dojde k mutaci genu, nejsou v průběhu života žádné klinické příznaky. Je extrémně vzácný.
- Rudimentární forma – latentní průběh se slabým projevem jednotlivých příznaků.
V závislosti na formě se stanoví prognóza a vypracuje se terapeutická taktika pro léčbu Huntingtonovy chorey.
Spektrum příznaků u Huntingtonovy chorey
Huntingtonova chorea vykazuje řadu příznaků, které mají tendenci se časem zhoršovat.
Zpočátku se na pozadí zdánlivé pohody mohou objevit mírné kognitivní poruchy – roztržitost, kolísání pozornosti, zapomnětlivost. Současně jsou zaznamenány jemné změny v charakteru a chování člověka: zvýšená podrážděnost, zášť, emoční nestabilita.
Brzy se objevují první motorické jevy v podobě prudkých, prudkých, mimovolních pohybů především v oblasti obličeje, krku a končetin – tzv. chorea. Současně dochází ke svalové rigiditě a pomalosti v jiných svalových skupinách.
Postupem času choreiformní hyperkineze postihuje stále více svalových skupin a šíří se po celém těle. Stále více trpí také kognitivně-mnestické funkce až do rozvoje výrazné demence v pozdních stádiích. Zároveň je pozorována celá řada závažných psychopatologických jevů – apatie, deprese, psychotické epizody.
Integrovaný přístup pro přesnou diagnostiku
Diagnostika Huntingtonovy chorey je založena na důkladném rozboru klinických projevů v kombinaci s laboratorním a instrumentálním vyšetřením.
Nejprve lékař shromáždí podrobnou anamnézu – zjišťuje, zda se v rodině nevyskytují podobná onemocnění, zda již dříve nebyly zaznamenány nějaké změny v osobnosti a chování. Poté podrobně zkoumá neurologický stav – posuzuje povahu mimovolní hyperkineze, možné kognitivní a emoční poruchy.
Pro objasnění diagnózy jsou předepsány instrumentální studie – CT nebo MRI mozku, které umožňují identifikovat atrofii určitých oblastí. Řada pacientů podstupuje DNA diagnostiku k detekci mutací v genu odpovědném za rozvoj onemocnění.
Komplexní rozbor získaných klinických, přístrojových a laboratorních dat dává lékaři možnost s vysokou mírou jistoty diagnostikovat nebo vyloučit Huntingtonovu choreu a také určit aktuální stadium onemocnění.
Kromě neurologického vyšetření, CT/MRI mozku a DNA diagnostiky mohou být provedeny další specializované testy k potvrzení Huntingtonovy chorey:
- Hodnocení motorických funkcí pomocí škál, jako je UHDRS, které kvantifikují závažnost chorey.
- Neuropsychologické testování, které identifikuje kognitivní poruchy pomocí standardizovaných metod.
- Nervové vedení a EP hodnocení studuje bioelektrickou aktivitu nervových vláken a jejich reakci na podněty.
- Ultrazvuk mozkových cév k vyloučení patologií napodobujících Huntingtonovu choreu.
- Biochemický krevní test k posouzení metabolismu a vyloučení jiných příčin hyperkineze.
Podrobné vyšetření nám umožňuje objasnit diagnózu a vytvořit personalizovaný program péče o pacienta.
Duševní poruchy u Huntingtonovy chorey
Progresivní demence je jedním z předních projevů onemocnění spolu s poruchami hybnosti.
Kromě toho pacienti zažívají širokou škálu psychopatologických jevů:
- Deprese
- Úzkostné poruchy;
- Apatie;
- Podrážděnost;
- Psychotické epizody.
Tyto poruchy výrazně zhoršují kvalitu života pacientů, a proto vyžadují adekvátní lékovou korekci.
Symptomatická a patogenetická terapie
Bohužel v současné době neexistuje žádná radikální léčba Huntingtonovy chorey. Aktivní patogenetická a symptomatická léčba však může zpomalit progresi onemocnění a zmírnit stav pacientů.
Na naší klinice je k léčbě pacientů s Huntingtonovou choreou využíván komplexní přístup, a to jak medikamentózní (psychofarmakoterapie), tak nemedikamentózními metodami.
Lékařské ošetření zahrnuje:
- Neuroprotektory pro ochranu nervových buněk.
- Anticholinergní léky ke snížení hyperkineze.
- Antidepresiva, trankvilizéry, neuroleptika pro korekci psychopatologických poruch.
Mezi používané nelékové metody patří:
- Kognitivní trénink pro zlepšení funkcí myšlení.
- Psychoterapie pro korekci emočních poruch.
V případě potřeby mohou být pacienti hospitalizováni na psychoneurologickém oddělení naší nemocnice k hloubkovému vyšetření, výběru adekvátní terapie a zmírnění exacerbací.
„Potýkat se s obtížemi? Vše je řešitelné! Přihlaste se k nám na konzultaci.”
Vedoucí lékařka T. L. Kharaburova
Přihlaste se ke konzultaci na telefonním čísle 8 (831) 266-03-06 nebo zanechte žádost. Přihlásit se
Poskytujeme celou řadu lékařských služeb pro diagnostiku, léčbu a rehabilitaci tohoto komplexního onemocnění:
- Konzultace se zkušenými neurology a psychiatry.
- Výběr účinné medikamentózní terapie.
- Kurzy kognitivního výcviku.
- Psychologická podpora pacientů a jejich příbuzných.
Neztrácejte naději! Společně můžeme s touto nemocí účinně bojovat.

Huntingtonova chorea je vzácné, dědičné onemocnění. Patologie postihuje nervový systém a je považována za nevyléčitelnou. Nemoc se z řečtiny překládá jako tanec – název je dán tím, že chorea se projevuje trhanými pohyby, které připomínají tanec. Stojí za to připomenout, že tato patologie není nezávislou chorobou, ale syndromem sestávajícím z příznaků, které jsou charakteristické pro určitá onemocnění.

Zkušenosti 33 let
Více než 27536 XNUMX konzultací
Abdrakhmanov Alexander Ravilievich
Terapeut, lékař rehabilitační medicíny
získejte konzultaci s 20% slevou
Bez registrace nebo instalace aplikace
959 ₽ 768 ₽

Abdrakhmanov Alexander Ravilievich
Získejte konzultaci
- Všeobecné informace
- Statistika
- Jak se nemoc přenáší?
- Příčiny
- Příznaky a stádia
- Etapy
- tvar
Všeobecné informace
Huntingtonova choroba postihuje striatum mozku a způsobuje choreickou hyperkinezi, kdy člověk vyvíjí trhavé, nekontrolované pohyby. Druhým, neméně častým názvem pro patologii je Svatovítský tanec. S progresí onemocnění se k poruchám hybnosti přidávají kognitivní poruchy, psychické poruchy a poruchy metabolismu.
Statistika
Od roku 2023 se onemocnění vyskytlo u 5 ze 100 000 lidí. Chorea je považována za nejčastější dědičnou patologii nervového systému. Nemoc je nerovnoměrně distribuována po celém světě: ve Venezuele je diagnostikována u 700 lidí ze 100 000, zatímco v afrických zemích se patologie nevyskytuje vůbec. První příznaky onemocnění se nejčastěji objevují až ve 40 letech, ale medicína zná případy, kdy nemoc debutovala v 70 letech a ve 20 letech.
Jak se nemoc přenáší?
Dědičnost Huntingtonovy choroby je autozomálně dominantní, což znamená, že existuje 50% pravděpodobnost, že je nemoc zděděna od jednoho z rodičů – patologie se přenáší rovnoměrně od otce i matky. Je těžké přesně říci, kolik dětí zdědí zmutovaný gen – genetici nedávají přesné předpovědi, ale pouze odhadují pravděpodobnost přenosu – nemoc se může přenést na jedno nebo více dětí, nebo nikdo neonemocní. Autosomálně dominantní mechanismus má v tomto případě vertikální dráhu, kdy je vertikální dráha onemocnění vysledována na rodokmenovém diagramu jedné rodiny, to znamená, že v každé generaci bude nemocný člověk. Pokud se ukáže, že celá rodina v některé generaci má čisté geny, přenos Huntingtonovy chorey se zastaví.
Nelze vyloučit falešné přeskočení generace, ke kterému dochází, když člověk předal mutovaný gen, ale zemřel dříve, než se objevily první příznaky onemocnění. U další generace se však symptomy stále vyvinou.
Neurologové a genetici identifikovali určité rysy dědičnosti poškozených genů. V každé další generaci se příznaky projeví dříve a průběh onemocnění bude závažnější. Taková statistika se potvrdí, když se mutovaný gen přenese od otce. Při vícenásobné dědičnosti po otci se u dětí v budoucích generacích nemoc brzy projeví. Například u dědečka se patologie projeví v 50 letech, u syna se rozvine v 35 letech a u vnuka ještě dříve – ve 14 letech.
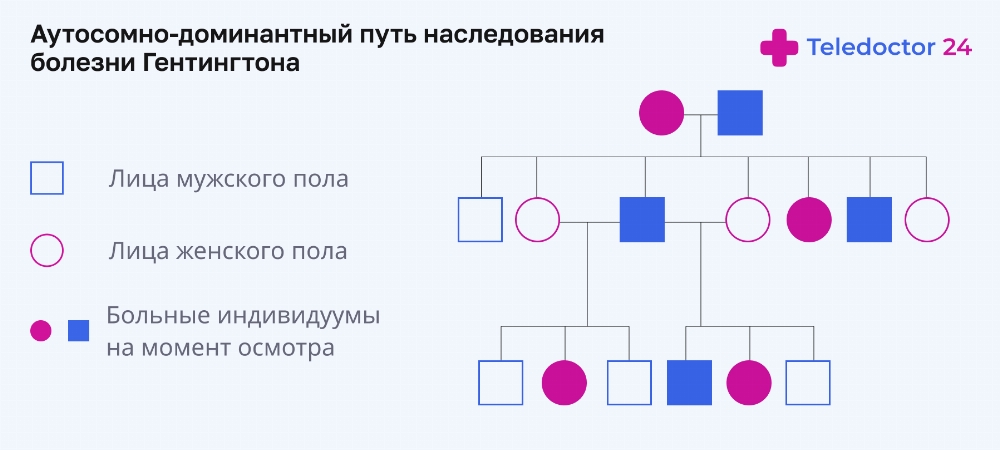
Foto: Rodokmen a dědičnost Huntingtonovy choroby
Příčiny
Je obtížné určit, co přesně způsobilo genovou mutaci, protože nemoc se dědí z generace na generaci. Hlavní příčinou onemocnění je narušení struktury určitých genů na 4. chromozomu nebo jejich mutace. Když dojde k mutacím a změní se složení nukleotidů (strukturní jednotka DNA) nebo kodonů (kombinace 3 nukleotidů, např. cytosin, adenin, guanin), změní se i genetický kód. V důsledku toho se začnou produkovat bílkoviny nesprávné struktury a někdy se jejich produkce úplně zastaví. V důsledku toho začnou buňky těla fungovat nesprávně. U Huntingtonovy choroby dochází k mutaci na 4. chromozomu, který kóduje syntézu speciální bílkoviny huntingtinu. Bohužel funkce tohoto proteinu není dosud plně objasněna. Předpokládá se, že huntingtin je normálně nezbytný pro tvorbu nervového systému – pomáhá produkovat další buněčné proteiny. Huntingtin se nachází ve velkém množství v mozku, ale nachází se i v jiných orgánech a tkáních. Při onemocnění je postižena ta část tohoto huntingtinu na samém počátku jeho proteinové struktury.
Podstatou mutace genu HTT je zvýšení počtu kopií CAG (CAG – cytosin, adenin, guanin). Pokud počet nukleotidových repetic přesáhne 35, pravděpodobnost genetické mutace se výrazně zvyšuje. V důsledku toho poškozený gen nadále produkuje protein s abnormální strukturou – huntingtin v tomto případě obsahuje nadbytek aminokyseliny glutaminu.
Důležité! Mezi ohrožené patří bezprostřední příbuzní pacienta. Mohou být přenašeči onemocnění a mají vysoké riziko onemocnění za předpokladu, že přežijí do doby klinických projevů. Ohroženy jsou také děti, bratři a sestry pacienta s choreou, protože jejich riziko zdědění mutace je více než 50 %.
Příznaky a stádia
Změněný protein se hromadí ve striatu mozku a v jeho kůře. Při jeho hromadění se během vývoje onemocnění rozvíjí dystrofie mozkových buněk, patologický proces postihuje i jiné části mozku.
Bohužel není možné odhalit příznaky onemocnění v raných stádiích, protože klinické příznaky se objeví blíže ke 40. roku věku – je to způsobeno dlouhým procesem akumulace abnormálních bílkovin (zvýšení množství bílkovin může trvat desítky let).
Když je postiženo striatum a subkortikální struktury, může pacient zaznamenat příznaky, jako jsou:
- prudké pohyby.
- mentálním postižením.
- mentální a kognitivní poruchy.
Na samém počátku onemocnění jsou příznaky chorey sotva patrné a člověk je stále dokáže ovládat. Záchvaty jsou nejčastěji náhodného charakteru – projevují se škubáním prstů, škubáním ramen, dupáním chodidly, škubáním nohou. Nejzřetelnějším příznakem je neschopnost udržet jazyk rovně – kdy pacient vyplázne jazyk a musí jej držet v natažené poloze bez pohybu po dobu 15 sekund. Jak nemoc postupuje, nekontrolované pohyby jsou častější a zapojují se nové svaly.
Hyperkineze má své typické vlastnosti. Oni:
- arytmické;
- spontánní;
- neuspořádaný;
- s různou amplitudou.
Pacientům se také mění chůze, stává se tancem, jejich řeč je narušena – když pacient mluví, je stále obtížnější mu rozumět. Mnoho lidí hlásí narušení funkce polykání v důsledku nucených pohybů svalů. Při emočním stresu, stejně jako při fyzické námaze, se také škubání zvyšuje. Během spánku se však mimovolní pohyby zastaví. Neustálé pohyby odebírají pacientovi mnoho energie, proto se nemoc vyznačuje zvýšenou únavou.
Mezi poruchy chování patří:
- kognitivní dysfunkce a odchylky;
- patologie spojené se sférou myšlení;
- problémy s pamětí.
Pacienti uvádějí, že si nepamatují materiál, který právě přečetli. Chorea ve většině případů ovlivňuje krátkodobou paměť a pozornost a pacient se nemůže soustředit. V tomto případě se ztrácí analytická funkce mozku a myšlení se stává primitivním. Člověk není schopen vidět abstraktně, ztrácí se schopnost logického myšlení.
Z psychologického hlediska mají pacienti řadu poruch:
- Postava se mění.
- Jádro osobnosti je zničeno.
- Objevují se odchylky v psychice.
- Emocionální a volní změny jsou zaznamenány.
Pacient se stává podrážděným, agresivním, vzteklým. Velmi často takoví lidé obviňují ostatní z výskytu této nemoci. Není vyloučeno neadekvátní, antisociální chování; jsou možné bludné stavy a halucinace. V ostatních případech je pacient letargický, uzavřený, apatický, ale deprese bude mít téměř každý.
Příznaky onemocnění se postupně zvyšují, takže pokud jste v rizikové skupině a u vašich blízkých příbuzných bylo toto onemocnění diagnostikováno, poraďte se s lékařem. Důležité je nejen onemocnění identifikovat a přesně stanovit diagnózu, ale také poskytnout psychologickou pomoc jak nemocnému, tak i jeho okolí. Naši lékaři jsou vám k dispozici 24/7.
